
In the last few days, there has been a near-panic reaction to the discovery of a mutation of the SARS-CoV-2 virus that causes COVID-19. The mutated lineage – in virology, a “lineage” suggests a variant somewhat closer to its immediate ancestors than a “strain” – is now named “Variant of Concern 202012/01,” having been referenced as “Variant Under Investigation 202012/01” and “B.1.1.7” earlier. (The VOC nomenclature is simply “year 2020, month 12, variant 01.)
Worries about the new variant focus on four main issues:
- Does it spread more easily?
- Does it cause more severe sickness or greater likelihood of death?
- Does it have resistance to vaccines just approved for use?
- Does it have the ability to escape detection by widely deployed PCR tests?
Mutations occur fairly often in the course of virus replication, essentially just inevitable errors making copies of copies of copies, but the vast majority have no meaningful effect. By random chance, very rarely a mutation gives an evolutionary reproductive advantage to the virus, which is how SARS-CoV-2 arose in the first place, probably many years ago in an animal reservoir, such as bats, until a spillover occurred to humans. Like every virus, this virus continues to mutate, and some random variations could boost the mutated lineage in epidemic risk. Thousands of such mutations have been observed and recorded in SARS-CoV-2; by one estimate about 12,000 distinct variants so far, but most do not seem to do anything different.
As the Global Initiative on Sharing Avian Influenza Data (GISAID) explained in a statement, “Mutations are naturally expected for viruses and are most often simply neutral regional markers useful for contact tracing. The changes seen have rarely affected viral fitness and almost never affected clinical outcome… Changes in the spike protein have relevance for potential effects on both host receptor as well as antibody binding with possible consequences for infectivity, transmission potential and antibody and vaccine escape. Actual effects need to be measured and verified experimentally.”
According to an advisory from the World Health Organization (WHO), by December 13 there were 1,108 known infections with this new variant in the UK. Ordinary COVID-19 testing is not intended to distinguish between variants, but in the UK between 5% and 10% of samples are routinely sent for genomic analysis that can distinguish between variants, and 4% of samples in southeastern England are sent for genomic analysis. The new variant accounts for over half of COVID-19 infections in southeastern England, after having been first seen in September in Kent, and is disproportionately more likely to be seen in young rather than old patients.
It is not clear why the variant is significantly more prevalent in the affected area, but one possible explanation is that the mutation makes the virus more transmissible from person to person, and if so this would be a major concern. The WHO warns, “Preliminary reports by the United Kingdom are that this variant is more transmissible than previous circulating viruses, with an estimated increase of between 40% and 70% in transmissibility (adding 0.4 to the basic reproduction number R0, bringing it to a range of 1.5 to 1.7).” On the other hand, according to a news article in Science, generally considered the most prestigious scientific journal in the US, “Scientists, meanwhile, are hard at work trying to figure out whether B.1.1.7 is really more adept at human-to-human transmission – not everyone is convinced yet – and if so, why.”
Sometimes increased prevalence is a result of a biological property of the virus, but this is not always so. It is widely known that the lineages of infections in the US toward the West Coast tend to be genetically closer to those found in Asia while the lineages of infections in the US toward the East Coast tend to be genetically closer to those found in Europe, but that almost certainly reflects patterns in airline travel rather than any intrinsic property of the mutations in the lineages. Likewise, the higher prevalence of the new variant in the UK could reflect human behavior, such as young people being more likely than old people to transmit infections in bars or restaurants, rather than any biological underpinning.
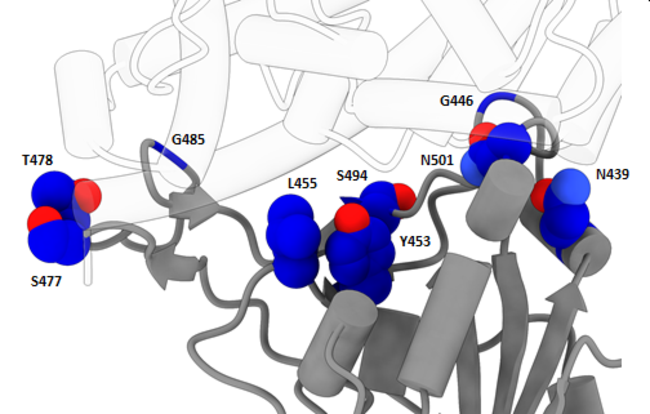
The new variant is characterized primarily by three distinct mutations, labeled “N501Y,” “P681H,” and “69-70del.” The detection of the new variant in the UK is a coincidence because a common commercial PCR test, known as TaqPath, consists of a three-part panel of subtests, one of which looks for the component of the virus that goes missing in the 69-70del mutation; as a result, a positive indication on the other two subtests and a negative indication on that subtest allowed the UK to go back over their existing test data and estimate the prevalence of the new variant.
While the 69-70del mutation can cause anomalous results in some PCR tests, it is the other two mutations that are of real concern, modifying the receptor-binding domain (RBD) that controls how the virus infects hosts cells. This could give the variant an advantage invading host cells, increasing ease of transmission from person to person, as the WHO fears. One indication of this possibility is that the N501Y mutation has been seen elsewhere in the world, with a few dozen cases known in Australia and Africa. In fact, it was the University of KwaZulu-Natal in South Africa that first identified the N501Y mutation in a separate lineage and made the UK aware of its potential importance. The independent evolution of this same mutation in different parts of the world strongly suggests that the mutation gives the virus some reproductive advantage and is therefore favored by natural selection. Whether this is true and if so to what extent is unknown pending further study.
So far, there is no evidence that the new variant causes more severe sickness or increased likelihood of death, even if it does have an advantage in being able to infect the host and therefore spread more easily. There is no known biological reason to expect a change in the RBD of the virus to result in worse sickness.
It’s too early to know whether the new variant is more resistant to immunity, whether acquired through recovered infection or vaccine, but theoretical models strongly suggest that the mechanisms of action by immune antibodies will be just as effective and are not affected by the particular mutations that characterize the new variant. Eventually, although SARS-CoV-2 mutates relatively slowly, it may be necessary to formulate a slightly modified vaccine periodically, similar to seasonal influenza vaccines that must be given annually.
Is the new variant in the US, or even in RI? Experts believe it almost certainly is, but there has been no testing for it until extremely recently, so it is impossible to know for certain. We asked that question, and Joseph Wendelken, spokesman for the RI Department of Health, told Motif, “Rhode Island participates in a CDC initiative to sequence SARS-CoV-2 from throughout the USA by regularly submitting positive specimens collected from Rhode Island residents. We are not aware of any such specimens being confirmed as the UK mutant strain. The State Health Laboratories (SHL) is also currently working with Rhode Island scientists on sequencing SARS-CoV-2 positive specimens at time points throughout the pandemic and in a sampling of isolates collected in March through August none were the UK mutant strain. These efforts are continuing.” However, as Wendelken made clear, at this point data more recent than August is not yet available, a month before the new variant was first detected in the UK. Motif asked the CDC directly, but so far they have not replied.
Wendelken was confident, however, that PCR testing accuracy in RI should not be affected if encountering the new variant. “Rhode Island clinical laboratories utilize a diversity of molecular amplification detection methods and many have multiple tests online – the SHL alone currently has five different ‘PCR’ tests. Each test uses different virus gene targets for detection and many use a multi-target approach. For this reason, if virus mutations in one area affect detection of a certain gene, the built-in redundancy would detect the other virus gene targets, thus lessening the potential for a ‘false negative’ result for the UK mutant strain.”